2.4 GS
It’s required to provide genotype and phenotype files to run GS in iPat. User-provided covariates will serve as fixed effects in the GS model, but none of GS methods will take user-defined kinship in iPat.
In iPat, users can perform two types of GS. If the ‘validation’ option is unchecked, iPat would estimate genetic estimated breeding values (GEBV) for samples in the whole dataset based on the phenotyped samples. Whereas when users check the option ‘Validation on accuracy’, iPat will split whole dataset into training and testing samples in order to verify the capability of trained GS model. For this manner, validation accuracies will be reported instead of GEBVs.
## Column 1 ['V1'] of item 2 is missing in item 1. Use fill=TRUE to fill with NA (NULL for list columns), or use.names=FALSE to ignore column names. use.names='check' (default from v1.12.2) emits this message and proceeds as if use.names=FALSE for backwards compatibility. See news item 5 in v1.12.2 for options to control this message.
## Column 1 ['V1'] of item 2 is missing in item 1. Use fill=TRUE to fill with NA (NULL for list columns), or use.names=FALSE to ignore column names. use.names='check' (default from v1.12.2) emits this message and proceeds as if use.names=FALSE for backwards compatibility. See news item 5 in v1.12.2 for options to control this message.
## Column 1 ['V1'] of item 2 is missing in item 1. Use fill=TRUE to fill with NA (NULL for list columns), or use.names=FALSE to ignore column names. use.names='check' (default from v1.12.2) emits this message and proceeds as if use.names=FALSE for backwards compatibility. See news item 5 in v1.12.2 for options to control this message.| Arguments | Default | Options | Description |
|---|---|---|---|
| Validation on accuracy | unchecked | checked, unchecked | If k-fold validation is performed in GS |
| Folds | 3 | 3, 5, 10 | K value for K-fold validation. For example, if k = 5, 20% samples will be tested as accuracy validation. |
| Iteration | 10 | Range from 10 to 100 | Number of iterations. Each iteration will split whole dataset into training part and testing part based on the specified k value for K-fold validation. |

Output files from GS include:
- Figures: GEBV distribution, Validation accuracies, Predicted versus observed values, Heterozygosity distribution, and Phenotype overview.
- Tables: GEBV report, Marker effects, Validation results, and Predicted values
2.4.1 gBLUP and rrBLUP
There’s no specific arguments available for both of these methods.
Reference
- Endelman,J. (2011) Ridge regression and other kernels for genomic selection in the R package rrBLUP. Plant Genome, 4, 250–255.
2.4.2 BGLR
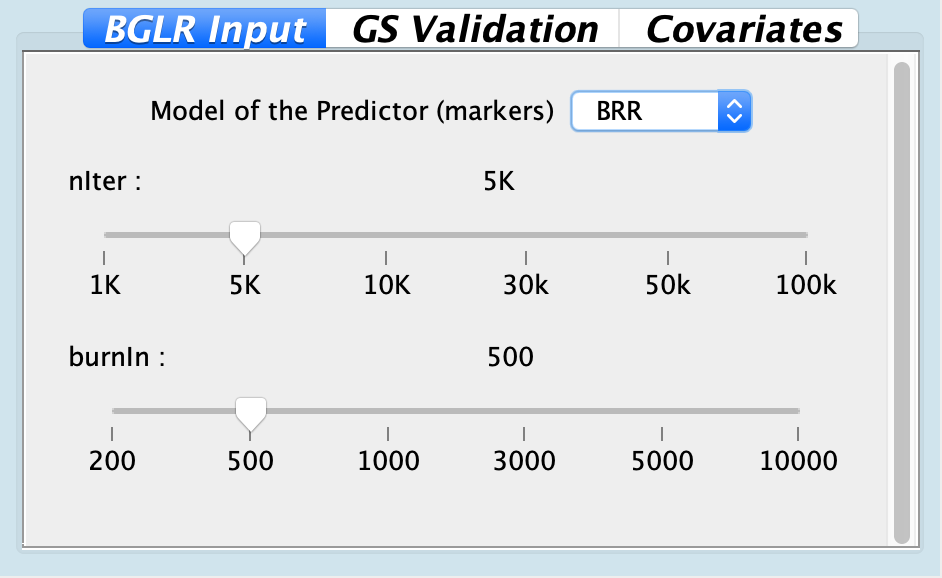
## Column 1 ['V1'] of item 2 is missing in item 1. Use fill=TRUE to fill with NA (NULL for list columns), or use.names=FALSE to ignore column names. use.names='check' (default from v1.12.2) emits this message and proceeds as if use.names=FALSE for backwards compatibility. See news item 5 in v1.12.2 for options to control this message.
## Column 1 ['V1'] of item 2 is missing in item 1. Use fill=TRUE to fill with NA (NULL for list columns), or use.names=FALSE to ignore column names. use.names='check' (default from v1.12.2) emits this message and proceeds as if use.names=FALSE for backwards compatibility. See news item 5 in v1.12.2 for options to control this message.
## Column 1 ['V1'] of item 2 is missing in item 1. Use fill=TRUE to fill with NA (NULL for list columns), or use.names=FALSE to ignore column names. use.names='check' (default from v1.12.2) emits this message and proceeds as if use.names=FALSE for backwards compatibility. See news item 5 in v1.12.2 for options to control this message.| Arguments | Default | Options | Description |
|---|---|---|---|
| Model for predictors | BRR | BRR, Bayes A, Bayes B, Bayes C, BL | Model to estimate marker effects |
| nIter | 5k | Range from 1k to 100k | Number of iteration |
| burnIn | 500 | Range from 200 to 10k | Burn-in values |
Reference
- Pérez, Paulino. et al. (2014) Genome-wide regression and prediction with the BGLR statistical package. Genetics, 198, 483-495.